Lab 3. Quantifying Biodiversity

This material must be reviewed by BCB743 students in Week 1 of Quantitative Ecology.
The seaweed (Smit et al. 2017) and toy data are at the links below:
- The seaweed species data –
SeaweedSpp.csv - The seaweed environmental data –
SeaweedEnv.csv - The seaweed coastal sections –
SeaweedSites.csv - The fictitious light data
light_levels.csv
Biodiversity The variability among living organisms from all sources including, inter alia, terrestrial, marine and other aquatic ecosystems and the ecological complexes of which they are part; this includes diversity within species, between species and of ecosystems.
— International Union for the Conservation of Nature (IUCN), Convention on Biological Diversity
The IUCN definition considers a diversity of diversity concepts. This module looks at diversity only at the species level (species diversity). However, we can also approach macroecological problems from phylogenetic and functional (and other) diversity concepts of view. Functional and phylogenetic diversity ideas will be introduced in the BDC743 module Quantitative Ecology.
Preparation
The South African Seaweed Data
In these examples, we will use the seaweed data of Smit et al. (2017). Please make sure that you read this paper. An additional file describing the background to the data is available here (Figure 1).
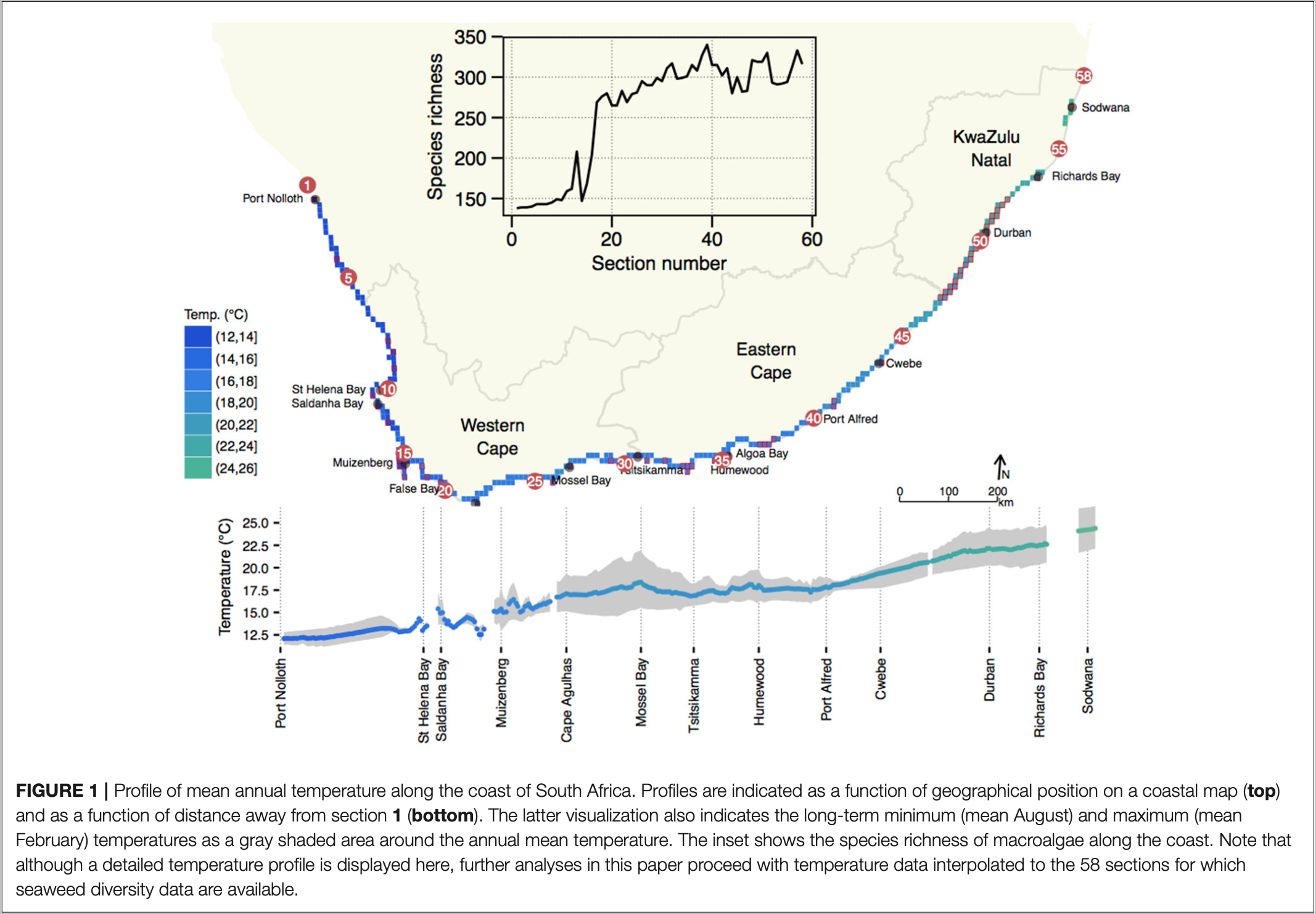
One of the datasets, SeaweedSpp.csv), comprises updated distribution records of 847 macroalgal species within each of 58 × 50 km-long sections of the South African coast (Bolton and Stegenga 2002). The dataset captures ca. 90% of the known seaweed flora of South Africa, but excludes some very small and/or very rare species for which data are insufficient. The data are from verifiable literature sources and John Bolton and Rob Anderson’s collections, assembled from information collected by teams of phycologists over three decades (Bolton 1986; Stegenga et al. 1997; Bolton and Stegenga 2002; De Clerck et al. 2005). Another file, env.csv), is a dataset of in situ coastal seawater temperatures derived from daily measurements over 40 years (Smit et al. 2013).
Setting Up the Analysis Environment
We will use R, so first, we must find, install and load various packages. Some packages will be available on CRAN and can be accessed and installed the usual way, but you will need to download others from R Forge.
A Look at the Data
Let’s load the data and see how it is structured:
spp <- read.csv('../data/seaweed/SeaweedSpp.csv')
spp <- dplyr::select(spp, -1)
# Lets look at the data:
dim(spp)[1] 58 847We see that our dataset has 58 rows and 847 columns. What is in the columns and rows? Start with the first five rows and five columns:
ACECAL ACEMOE ACRVIR AROSP1 ANAWRI
1 0 0 0 0 0
2 0 0 0 0 0
3 0 0 0 0 0
4 0 0 0 0 0
5 0 0 0 0 0Now the last five rows and five columns:
WOMKWA WOMPAC WRAARG WRAPUR WURMIN ZONSEM
53 0 0 1 0 0 0
54 0 0 1 0 0 0
55 0 0 1 0 0 0
56 0 1 1 0 1 0
57 1 0 1 0 1 0
58 0 0 1 0 1 0So, each row corresponds to a site (i.e. each of the coastal sections), and each column contains a species. We arrange the species alphabetically and use a six-letter code to identify them.
Species Data
When ecologists talk about species diversity, they typically consider the characteristics of biological communities in a specific habitat, ecological community, or ecosystem. Species diversity considers three essential concepts about how species are distributed in space: their richness, abundance, and evenness. We can express each of these as biodiversity metrics that allow us to compare communities in space and time.
When ecologists talk about ‘biodiversity’, they might not necessarily be interested in all the plants and animals and things that are neither plant nor animal that occur at a particular place. Some ecologists are interested in ants and moths. Others might find fish more insightful. Some even like marine mammals! I prefer seaweed. The analysis of biodiversity data might often be constrained to some higher-level taxon, such as all angiosperms in a landscape, reptiles, etc. (but we sample all species in the higher-level taxon). Some ecological questions benefit from comparisons of diversity assessments among selected taxa (avifauna vs small mammals, for example), as this focus might address some particular ecological hypothesis. The bird vs small mammal comparison might reveal how barriers such as streams and rivers structure biodiversity patterns. In our examples, we will use such focused datasets.
Here we look at the various measures of biodiversity, viz.
Three Measures of Biodiversity:
Whittaker (1972) coined three measures of biodiversity, and the concepts were ‘modernised’ by Jurasinski et al. (2009). The concepts represent the measurement of biodiversity across different spatial scales.
By now, you will have received a brief Introduction to R, and we can proceed with looking at some of the measures of biodiversity. We will start by using data on the seaweeds of South Africa to demonstrate some ideas around diversity measures. The vegan1 (for vegetation analysis) package (Oksanen et al. 2022) offers various functions to calculate diversity indices. I will demonstrate some of these functions below.
1 I am by no means an advocate for veganism.
Alpha-Diversity
We can represent
- as species richness,
- as a univariate diversity index, such as the
- Species evenness, e.g. Pielou’s evenness,
We will work through each in turn.
Species Richness,
First, is species richness, which we denote by the symbol
In the seaweed biodiversity data, I count the number of species within each of the sections. This is because we view each coastal section as the local scale (the smallest unit of sampling).
The preferred option for calculating species richness is the specnumber() function in vegan:
1specnumber(spp, MARGIN = 1)- 1
-
The
MARGIN = 1argument tells R to calculate the number of species within each row (site).
[1] 138 139 139 140 143 143 143 145 149 148 159 162 208 147 168 204 269 276 280
[20] 265 265 283 269 279 281 295 290 290 299 295 311 317 298 299 301 315 308 327
[39] 340 315 315 302 311 280 300 282 283 321 319 319 330 293 291 292 294 313 333
[58] 316The data output is easier to understand if we display it as a tibble():
# A tibble: 6 × 2
section richness
<int> <int>
1 1 138
2 2 139
3 3 139
4 4 140
5 5 143
6 6 143The diversityresult() function in the BiodiversityR package can do the same (sometimes this package is difficult to install due to various software dependencies that might be required for the package to load properly—do not be sad if this method does not work):
Now we make a plot seen in Figure 2:

In other instances, it makes more sense to calculate the mean species richness of all the sampling units (e.g. quadrats) taken inside the ecosystem of interest. How you calculate and present species richness depend on your research question and so you will have to decide based on your data and study.
In the seaweed study, the mean ± SD species richness across all of the 58 coastal sections is:
Univariate Diversity Indices
The second way we can express
Shannon’s
It is calculated as:
Simpson’s
Fisher’s fisher.alpha() and fisherfit()). The estimation is possible only for actual counts (i.e. integers) of individuals, so it will not work for per cent cover, biomass, and other measures that real numbers can express. It’s especially useful for comparing the diversity of samples with different total abundances. We will get to this function later under Fisher’s logarithmic series.
Except for Fisher’s-
Site A B C D E F
1 low_light 0.75 0.62 0.24 0.33 0.21 0.14
2 mid_light 0.38 0.15 0.52 0.57 0.28 0.29
3 high_light 0.08 0.15 0.18 0.52 0.54 0.56We can see above that instead of having data with 1s and 0s for presence-absence, here we have some values that indicate the relative number of individuals belonging to each of the species in the three light environments. We calculate species richness (as before), and also the Shannon and Simpson indices using vegan’s diversity() function:
light_div <- tibble(
site = c("low_light", "mid_light", "high_light"),
richness = specnumber(light[, 2:7], MARGIN = 1),
shannon = round(diversity(light[, 2:7], MARGIN = 1, index = "shannon"), 2),
simpson = round(diversity(light[, 2:7], MARGIN = 1, index = "simpson"), 2)
)
light_div# A tibble: 3 × 4
site richness shannon simpson
<chr> <int> <dbl> <dbl>
1 low_light 6 1.62 0.78
2 mid_light 6 1.71 0.81
3 high_light 6 1.59 0.77Evenness refers to the shape of a species abundance distribution, which suggests the relative abundance of different species.
One index for evenness is Pielou’s evenness,
where
To calculate Pielou’s evenness index for the light data, we can do this:
H <- diversity(light[, 2:7], MARGIN = 1, index = "shannon")
J <- H/log(specnumber(light[, 2:7]))
round(J, 2)[1] 0.91 0.95 0.89Berger-Parker Index indicates the proportion of the community that the most abundant species represents. It is given by the formula:
Chao1 and ACE are estimators often used to predict the total species richness in a community based on the number of rare species observed in samples.
Gamma-Diversity
Returning to the seaweed data,
We can also use:
(To be reviewed by BCB743 student but not for marks)
- Why is there a difference between the two?
- Which is correct?
Think before you calculate
Beta-Diversity
Whittaker’s
The first measure of
true_beta <- data.frame(
beta = specnumber(spp, MARGIN = 1) / ncol(spp),
section_no = c(1:58)
)
# true_beta
ggplot(data = true_beta, (aes(x = section_no, y = beta))) +
geom_line(size = 1.2, colour = "indianred") +
xlab("Coastal section, west to east") +
ylab("True beta-diversity") +
theme_linedraw()
The second measure of
abs_beta <- data.frame(
beta = ncol(spp) - specnumber(spp, MARGIN = 1),
section_no = c(1:58)
)
# abs_beta
ggplot(data = abs_beta, (aes(x = section_no, y = beta))) +
geom_line(size = 1.2, colour = "indianred") +
xlab("Coastal section, west to east") +
ylab("Absolute beta-diversity") +
theme_linedraw()
Contemporary Definitions
Contemporary definitions of ?vegdist. However, discussing pairwise dissimilarities with
Dissimilarity indices
Dissimilarity indices are special cases of diversity indices that use pairwise comparisons between sampling units, habitats, or ecosystems.
Species dissimilarities result in pairwise matrices similar to the pairwise correlation or Euclidian distance matrices we have seen in Lab 1. In Lab 2b you will have also learned how to calculate these ecological distances in R. These dissimilarity indices are multivariate and compare between sites, sections, plots, etc., and must therefore not be confused with the univariate diversity indices.
We use the Bray-Curtis and Jaccard indices with abundance data and the Sørensen dissimilarity with presence-absence data. The seaweed dataset is a presence-absence dataset that necessitates using the Sørensen index. The interpretation of the resulting square (number of rows = number of columns) dissimilarity matrices is the same regardless of whether we calculate it for an abundance or presence-absence dataset. The values in the matrix range from 0 to 1. A 0 means that the pair of sites we compare is identical (all species in common) but 1 means they are completely different (no species in common). In the square dissimilarity matrix, the diagonal is 0, which essentially (and obviously) means that any site is identical to itself. Elsewhere the values will range from 0 to 1. Since this is a pairwise calculation (each site compared to every other site), our seaweed dataset will contain (58 × (58 - 1))/2 = 1653 values, each one ranging from 0 to 1.
The first step involves the species table,
The vegan function vegdist() provides access to the dissimilarity indices. We calculate the Sørensen dissimilarity index:
sor <- vegdist(spp, binary = TRUE) # makes the lower triangle matrix
sor_df <- round(as.matrix(sor), 4)
dim(sor_df)[1] 58 58 1 2 3 4 5 6 7 8 9 10
1 0.0000 0.0036 0.0036 0.0072 0.0249 0.0391 0.0391 0.0459 0.0592 0.0629
2 0.0036 0.0000 0.0000 0.0036 0.0213 0.0355 0.0355 0.0423 0.0556 0.0592
3 0.0036 0.0000 0.0000 0.0036 0.0213 0.0355 0.0355 0.0423 0.0556 0.0592
4 0.0072 0.0036 0.0036 0.0000 0.0177 0.0318 0.0318 0.0386 0.0519 0.0556
5 0.0249 0.0213 0.0213 0.0177 0.0000 0.0140 0.0140 0.0208 0.0342 0.0378
6 0.0391 0.0355 0.0355 0.0318 0.0140 0.0000 0.0000 0.0069 0.0205 0.0241
7 0.0391 0.0355 0.0355 0.0318 0.0140 0.0000 0.0000 0.0069 0.0205 0.0241
8 0.0459 0.0423 0.0423 0.0386 0.0208 0.0069 0.0069 0.0000 0.0136 0.0171
9 0.0592 0.0556 0.0556 0.0519 0.0342 0.0205 0.0205 0.0136 0.0000 0.0034
10 0.0629 0.0592 0.0592 0.0556 0.0378 0.0241 0.0241 0.0171 0.0034 0.0000What we see above is a square dissimilarity matrix. The most important characteristics of the matrix are:
- whereas the raw species data,
- the diagonal is filled with 0;
- the matrix is symmetrical—it is comprised of symetrical upper and lower triangles.
Create a data.frame suitable for plotting:
(To be reviewed by BCB743 student but not for marks)
These questions concern matrices produced from species data using any of the indices available in vegdist():
- Why is the matrix square, and what determines the number of rows/columns?
- What is the meaning of the diagonal?
- What is the meaning of the non-diagonal elements?
- Referring to the seaweed species data specifically, take the data in row 1 or column 1 and create a line graph showing these values as a function of the section number.
- Provide a mechanistic (ecological) explanation for why this figure takes the shape that it does. Which community assembly process does this hint at?
There are different interpretations linked to
Species turnover and nestedness-resultant
There are two kinds of
How do we calculate the turnover and nestedness-resultant components of betapart.core() and betapart.pair() functions. The outcomes of this partitioning calculation are placed into the matrices
# Decompose total Sørensen dissimilarity into turnover and nestedness-resultant
# components:
Y.core <- betapart.core(spp)
Y.pair <- beta.pair(Y.core, index.family = "sor")
# Let Y1 be the turnover component (beta-sim):
Y1 <- data.frame(round(as.matrix(Y.pair$beta.sim), 3))
# Let Y2 be the nestedness-resultant component (beta-sne):
Y2 <- data.frame(round(as.matrix(Y.pair$beta.sne), 3))A portion of the turnover component matrix:
X1 X2 X3 X4 X5 X6 X7 X8 X9 X10
1 0.000 0.000 0.000 0.000 0.007 0.022 0.022 0.022 0.022 0.029
2 0.000 0.000 0.000 0.000 0.007 0.022 0.022 0.022 0.022 0.029
3 0.000 0.000 0.000 0.000 0.007 0.022 0.022 0.022 0.022 0.029
4 0.000 0.000 0.000 0.000 0.007 0.021 0.021 0.021 0.021 0.029
5 0.007 0.007 0.007 0.007 0.000 0.014 0.014 0.014 0.014 0.021
6 0.022 0.022 0.022 0.021 0.014 0.000 0.000 0.000 0.000 0.007
7 0.022 0.022 0.022 0.021 0.014 0.000 0.000 0.000 0.000 0.007
8 0.022 0.022 0.022 0.021 0.014 0.000 0.000 0.000 0.000 0.007
9 0.022 0.022 0.022 0.021 0.014 0.000 0.000 0.000 0.000 0.000
10 0.029 0.029 0.029 0.029 0.021 0.007 0.007 0.007 0.000 0.000A portion of the nestedness-resultant matrix:
X1 X2 X3 X4 X5 X6 X7 X8 X9 X10
1 0.000 0.004 0.004 0.007 0.018 0.017 0.017 0.024 0.037 0.034
2 0.004 0.000 0.000 0.004 0.014 0.014 0.014 0.021 0.034 0.030
3 0.004 0.000 0.000 0.004 0.014 0.014 0.014 0.021 0.034 0.030
4 0.007 0.004 0.004 0.000 0.011 0.010 0.010 0.017 0.030 0.027
5 0.018 0.014 0.014 0.011 0.000 0.000 0.000 0.007 0.020 0.017
6 0.017 0.014 0.014 0.010 0.000 0.000 0.000 0.007 0.021 0.017
7 0.017 0.014 0.014 0.010 0.000 0.000 0.000 0.007 0.021 0.017
8 0.024 0.021 0.021 0.017 0.007 0.007 0.007 0.000 0.014 0.010
9 0.037 0.034 0.034 0.030 0.020 0.021 0.021 0.014 0.000 0.003
10 0.034 0.030 0.030 0.027 0.017 0.017 0.017 0.010 0.003 0.000A portion of the nestedness-resultant matrix reformatted as a tibble()2:
2 Note that the rows are no longer numbered in the tibble view, but it can easily be recreated by seq(1:58).
# A tibble: 6 × 58
X1 X2 X3 X4 X5 X6 X7 X8 X9 X10 X11 X12 X13
<dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 0 0.004 0.004 0.007 0.018 0.017 0.017 0.024 0.037 0.034 0.069 0.078 0.196
2 0.004 0 0 0.004 0.014 0.014 0.014 0.021 0.034 0.03 0.065 0.074 0.193
3 0.004 0 0 0.004 0.014 0.014 0.014 0.021 0.034 0.03 0.065 0.074 0.193
4 0.007 0.004 0.004 0 0.011 0.01 0.01 0.017 0.03 0.027 0.062 0.071 0.19
5 0.018 0.014 0.014 0.011 0 0 0 0.007 0.02 0.017 0.052 0.061 0.181
6 0.017 0.014 0.014 0.01 0 0 0 0.007 0.021 0.017 0.053 0.062 0.184
# ℹ 45 more variables: X14 <dbl>, X15 <dbl>, X16 <dbl>, X17 <dbl>, X18 <dbl>,
# X19 <dbl>, X20 <dbl>, X21 <dbl>, X22 <dbl>, X23 <dbl>, X24 <dbl>,
# X25 <dbl>, X26 <dbl>, X27 <dbl>, X28 <dbl>, X29 <dbl>, X30 <dbl>,
# X31 <dbl>, X32 <dbl>, X33 <dbl>, X34 <dbl>, X35 <dbl>, X36 <dbl>,
# X37 <dbl>, X38 <dbl>, X39 <dbl>, X40 <dbl>, X41 <dbl>, X42 <dbl>,
# X43 <dbl>, X44 <dbl>, X45 <dbl>, X46 <dbl>, X47 <dbl>, X48 <dbl>,
# X49 <dbl>, X50 <dbl>, X51 <dbl>, X52 <dbl>, X53 <dbl>, X54 <dbl>, …(To be reviewed by BCB743 student but not for marks)
- Plot species turnover as a function of Section number, and provide a mechanistic explanation for the pattern observed.
- Based on an assessment of literature on the topic, provide a discussion of nestedness-resultant
The Lab 3 assignment is due at 07:00 on Monday 12 August 2022.
Provide a neat and thoroughly annotated R file which can recreate all the graphs and all calculations. Written answers must be typed in the same file as comments.
Please label the R file as follows:
BDC334_<first_name>_<last_name>_Lab_3.R
(the < and > must be omitted as they are used in the example as field indicators only).
Submit your appropriately named R documents on iKamva when ready.
Failing to follow these instructions carefully, precisely, and thoroughly will cause you to lose marks, which could cause a significant drop in your score as formatting counts for 15% of the final mark (out of 100%).
References
Reuse
Citation
@online{smit,_a._j.2021,
author = {Smit, A. J.,},
title = {Lab 3. {Quantifying} {Biodiversity}},
date = {2021-01-01},
url = {http://tangledbank.netlify.app/BDC334/03-biodiversity1.html},
langid = {en}
}